



Коллектив авторов
Внутренние болезни. Том 2
Альтернативным подходом к лечению острых форм ИТ является внутривенное (в течение 5 дней) введение больших доз (3 г) хорошо очищенного иммуноглобулина G, который особенно важен у детей и беременных.
Второй «линией обороны» хронической ИТ являются антилимфоцитарный глобулин и/или спленэктомия. Основными показаниями для спленэктомии считаются:
– недостаточная эффективность ранее проводимой терапии глюкокортикоидами, в том числе в комбинации с иммунодепрессантами;
– ограниченные возможности длительной терапии глюкокортикоидами у больных сахарным диабетом, язвенной болезнью, артериальной гипертензией, остеопорозом и некоторыми другими нарушениями обмена;
– способ профилактики будущих катастроф у женщин с ИТ, намеревающихся рожать детей.
Спленэктомия проводится в условиях специализированного хирургического стационара под контролем гемограммы и уровня тромбоцитов. Для предотвращения развития нежелательной надпочечниковой недостаточности плановая доза глюкокортикоидов непосредственно перед операцией должна удвоиться. Кроме того, из-за высокого риска развития пневмококкового сепсиса у спленэктомированных больных все готовящиеся к спленэктомии пациенты должны быть предварительно иммунизированы пневмококковыми вакцинами.
В результате такой тактики ведения больных ИТ стойкого положительного эффекта удается достичь у 90 % детей и приблизительно у 60 – 70 % взрослых. Из других способов лечения, используемых для лечения рефрактерных к стандартной терапии ИТ, заслуживают внимания: а) большие дозы метилпреднизолона; б) иммуносупрессивная терапия циклофосфаном или азатиоприном; в) винкристин, винбластин, колхицин или этопозид; г) низкие дозы антирезус-0 (D) иммуноглобулина G; д) даназол; е) á-интерферон; е) аферез или плазмаферез с антистафилококковым протеином А, которые направлены на удаление из циркуляции антитромбоцитарных антител; ж) большие дозы аскорбиновой кислоты; з) дапсон; и) лечебное анти-СD20-антитело – мабтера; к) ромипластин.
Механизм действия при ИТ алкалоидов барвинка розового (винкристина и винбластина), колхицина и группы алкалоидов подофиллотоксина (вепезид и этопозид) приблизительно одинаков. Он связан с прямым воздействием указанных препаратов на основной опорный белок цитоскелета тромбоцитов и макрофагов – тубулин. Поскольку последний формирует микротрубочки, без которых полноценный фагоцитоз невозможен, эта функция макрофагов во время лечения названными препаратами частично утрачивается, в том числе и в отношении покрытых антителами тромбоцитов.
Обычно винкристин для лечения ИТ вводится внутривенно в дозе 0,5 мг один раз в неделю, и такая терапия продолжается в течение 6 нед. Эффективность терапии оценивают через 2 – 3 нед., а в случае успеха эффект может сохраняться 6 мес. и более. До недавнего времени основным сигналом для остановки терапии винкристином являлась периферическая нейропатия в виде полинейропатии, локальных невритов и парезов, в особенности кишечника, частота которых при использовании вышеупомянутых малых доз винкристина стала минимальной.
Колхицин, подобно винкристину и винбластину, связывается с тубулином и нарушает такие зависимые от микротрубочек процессы в макрофагально-мононуклеарной системе, как фагоцитоз. В результате его применения полные и частичные ремиссии ИТ удается получить у 20 и 40 % больных соответственно.
Что касается этопозида, этот препарат может обеспечить длительные стойкие ремиссии у больных с недавно возникшей ИТ. При этом ожидаемого миелотоксического эффекта от действия этопозида не наблюдалось.
Внутривенные инфузии иммуноглобулина G в дозе 400 мг/кг/сут, вводимые в течение 5 дней, способны поднять уровень тромбоцитов крови выше 50 % 109/л у 83 % леченых пациентов. Одним из объяснений положительного эффекта иммуноглобулина при ИТ является блокада Fc-рецептора макрофагов и отсюда меньшее повреждающее воздействие последних на циркулирующие в крови тромбоциты.
Основными же недостатками терапии большими дозами иммуноглобулина G следует считать ее дороговизну и нестойкость эффекта. Поэтому назначение высоких доз иммуноглобулина G, прежде всего, оправданно для купирования остро возникающего геморрагического синдрома у детей, а также как важный этап подготовки к спленэктомии.
Альтернативным подходом для блокады Fс-рецептора макрофагальномононуклеарной системы у Rh-позитивных больных с ИТ может быть внутривенное или внутримышечное введение анти-Rho иммуноглобулина G в дозах 0,75 – 4,5 мг в течение 1 – 5 дней.
Даназол представляет собой алкилированный в 17-й позиции синтетический андроген с минимальным вирилизирующим эффектом. Клинический опыт показывает, что этот эффект даназола при ИТ во многом зависит от длительности терапии, возраста и пола больных, а также от состояния их селезенки. В частности, женщины реагируют на даназол лучше мужчин. В то же время ранее спленэктомированные больные (50 – 60 %) на даназол практически не реагируют. Иммуномодулирующий эффект даназола при ИТ связывают с уменьшением продукции антитромбоцитарных антител, а также с уменьшением числа Fс-(IgG-) рецепторов на фагоцитирующих тромбоциты клетках. Рекомендуемые начальные дозы даназола должны быть не менее 600 – 800 мг/сут, в то время как для поддержания эффекта достаточно приема 200 мг два раза в неделю.
Положительный эффект á-интерферона при лечении взрослых больных с ИТ связывают с его прямым ингибирующим влиянием на В-клетки. Обычно для получения эффекта он вводится в дозе 3 млн ед. в течение 12 дней. Основные побочные эффекты интерферона – различные аллергические реакции, лихорадка разной степени выраженности, а главное – утяжеление самой тромбоцитопении (из-за его прямого тормозящего влияния на деление костномозговых элементов).
Из других способов терапии ИТ заслуживают внимания: а) ежедневная очистка плазмы от антитромбоцитарных антител; б) удаление антитромбоцитарных антител с помощью плазмафереза со стафилококковым протеином-А; в) использование больших доз аскорбиновой кислоты, вводимых внутривенно; г) применение дапсона; д) внутривенное введение анти-СD20-антитела – мабтеры; е) использование агониста тромбоцитарного рецептора ромипластина. Два последних препарата дороги. Однако положительный опыт использования их при тяжелых, рефрактерных к стандартной терапии формах ИТ, в том числе после аллоТГСК, обнадеживает.
5.6.2. Тромбоцитопатии
Определение. Тромбоцитопатии – это врожденные нарушения тромбоцитарного звена гемостаза, связанные с продукцией костным мозгом неполноценных в отношении адгезии, агрегации или реакции высвобождения (секреции) пластинок. Они наследуются по аутосомно-доминантному или рецессивному типу и имеют однотипную клиническую картину.
Нарушение адгезии тромбоцитов к субэндотелиальным элементам имеет место при болезни Бернара – Сулье. В основе нарушения гемостаза лежит дефект тромбоцитарной мембраны, а именно отсутствие на ней гликопротеина Ib, что приводит к нарушению взаимодействия тромбоцитов с ФВ (см. цв. вкл., рис. 5.14), а через него – с субэндотелиальными структурами сосудистой стенки. Примером заболевания с нарушением фазы первичной агрегации тромбоцитов может быть тромбастения Гланцмана, при которой главную роль в патогенезе нарушений тромбоцитарного звена гемостаза играет дефицит/дефект мембранного гликопротеинового комплекса IIb/IIIa, являющегося рецептором для фибриногена. Взаимодействие последнего с гликопротеином IIb/IIIa и Са2+ необходимо для агрегации тромбоцитов и ретракции кровяного сгустка. В итоге тромбоциты больных тромбастенией Гланцмана не способны реагировать полноценно ни на один из стимуляторов агрегации, в том числе на АДФ, адреналин и коллаген. При этом имеет место также отчетливое снижение ретракции кровяного сгустка.
Клиническая картина. Геморрагические проявления при наследственных тромбоцитопатиях появляются в раннем детстве и имеют однотипный характер. Доминируют петехии и пятнистые кровоизлияния на коже. Они появляются спонтанно или после небольших травм. Характерны также десневые, носовые и маточные кровотечения. Реже встречаются желудочно-кишечные кровотечения и кровотечения во время операций и родов.
Диагноз. Мысль о врожденной тромбоцитопатии возникает у врача при наличии в клинике кровоточивости капиллярного типа, наличии удлиненной кровоточивости на фоне нормального содержания тромбоцитов в крови. Лабораторное обследование позволяет выявить первичное нарушение адгезии, агрегации или секреции в тромбоцитах, которое может сопровождаться также нарушением ретракции кровяного сгустка.
Дифференциальный диагноз тромбоцитопатий следует проводить с другими геморрагическими диатезами, проявляющими себя нарушением функции тромбоцитов, в частности с болезнью Виллебранда. В отличие от тромбастении Гланцмана, при которой нарушены все виды агрегации тромбоцитов, приболезни Виллебранда прежде всего нарушается реакция на ристомицин (ристоцитин), которая корригируется добавлением ФВ. В отличие же от болезни Виллебранда при синдроме Бернара – Сулье добавление ФВ не корригирует выявленный дефект нарушенной агрегации тромбоцитов на ристоцитин, поскольку он связан с отсутствием на тромбоцитах Ib гликопротеинового рецептора для ФВ. Кроме того, при болезни Виллебранда, помимо нарушения в адгезии тромбоцитов, наблюдается снижение коагуляционной активности фактора VIII (VIII: C), поскольку в норме ФВ выполняет роль носителя коагуляционного компонента фактора VIII и защищает его от разрушения в крови. Наконец, при синдроме Бернара – Сулье в мазках крови могут присутствовать гигантские тромбоциты, что для болезни Виллебранда не характерно.
Лечение. Некоторое улучшение функциональной активности тромбоцитов при кровотечении может быть достигнуто с помощью дицинона (внутривенно, per os) или 1 % раствора АТФ (по 1,0 г внутримышечно ежедневно) со жженой магнезией по 0,5 г 3 раза в сутки в течение месяца или внутримышечно 10 инъекций адроксона (по 1 мл 2 – 4 раза в день). Положительный эффект приносит фитотерапия (настои крапивы, тысячелистника и подорожника).
Прогноз при наследственных тромбоцитопатиях, как правило, благоприятный. Естественно, он ухудшается при кровоизлияниях в мозг и в сетчатку глаз, приводящих к церебральным нарушениям и к потере зрения.
5.6.3. Вазопатии
Геморрагические диатезы, связанные с поражением сосудов, могут иметь врожденный и приобретенный характер.
Наследственные геморрагические вазопатии включают болезнь Ослера, синдромы Черногубова – Элерса – Данлоса и Марфана, а также гемангиомы. Их патогенез так или иначе связан с неполноценностью, точнее с неправильным развитием соединительной ткани, в том числе субэндотелия сосудов. Кровоточивость обусловлена малой резистентностью и легкой ранимостью сосудистой стенки, в частности в местах телеангиэктазий, а также слабой стимуляцией в этих участках адгезии и агрегации тромбоцитов.
Кровоточивость при наследственных вазопатиях обнаруживает себя в детском возрасте. Речь идет о легко возникающих геморрагиях в кожу, кровотечениях из слизистых оболочек носа, ротовой полости, желудочно-кишечного тракта. Характерны кровотечения из десен во время чистки зубов. Опасны кровоизлияния в мозг и внутренние органы.
Диагноз ставится на основании рано возникшего геморрагического диатеза у больных с возможными дефектами соединительной ткани, проявляющими себя различными пороками развития, но при отсутствии каких-либо выраженных изменений в тромбоцитарном звене гемостаза. В качестве вспомогательного разграничивающего теста может быть использован аспирин, при назначении которого (0,5 г) у больных с вазопатиями геморрагический диатез должен усиливаться, а при тромбоцитопениях нарастает незначительно.
Лечение наследственных вазопатий носит симптоматический характер. Для остановки кровотечения используют гемостатическую губку, фибриноген, прижигание кровоточащих сосудов и т. д.
Прогноз для жизни относительно благоприятный, хотя оснований для беспокойства у таких пациентов в жизни предостаточно.
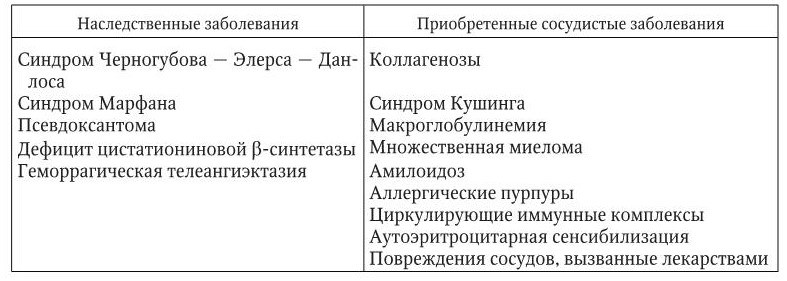
Приобретенные вазопатии с проявлениями геморрагического диатеза встречаются в клинике значительно чаще врожденных (табл. 5.3), хотя в клиническом плане мало отличаются от них.
Основные лекарства, вызывающие вазопатии: аспирин, йод; изониазид, мепробамат, метилдопа, пиперазин, хинидин, гипотиазид, мышьяк, толбутамид, аллопуринол, резерпин, индометацин, соли золота, фуросемид, эстрогены, дигоксин, хлорпропамид, хинин, сульфаниламиды, левомицетин, варфарин и др.
Диагноз приобретенных вазопатий ставится при наличии в клинике разной степени выраженности поражений сосудов (атеросклероз, диабетическая ангиопатия, системные васкулиты и др.) в отсутствие отмеченных выше нарушений тромбоцитарного звена гемостаза.
Лечение направлено на коррекцию основного заболевания и неспецифическое устранение симптомов геморрагического диатеза.
Таблица 5.3
Заболевания сосудов, проявляющиеся повышенной кровоточивостью
5.6.4. Коагулопатии
Определение. Коагулопатиями называются геморрагические диатезы, связанные с нарушением плазменного звена гемостаза. Они носят или врожденный (болезнь Виллебранда, гемофилии), или приобретенный характер (ДВС-синдром).
Болезнь Виллебранда
Болезнь Виллебранда является самой распространенной формой среди наследственных геморрагических диатезов. Заболевание передается по аутосомно-доминантному или рецессивному типу и встречается как у мужчин, так и у женщин. Поскольку ФВ необходим для полноценной адгезии и агрегации тромбоцитов и для поддержания антигемофильного глобулина, его недостаток приводит к нарушению как сосудисто-тромбоцитарного, так и плазменного звеньев гемостаза. В свою очередь, наличие двойного дефекта в системе гемостаза приводит к тому, что болезнь Виллебранда, с одной стороны, напоминает тромбоцитопатию, с другой – гемофилию.
Патогенез. Фактор Виллебранда представляет собой гликопротеин с молекулярной массой от 500 до 12 000 – 20 000 кДа (см. цв. вкл., рис. 5.13). Он состоит из однотипных димерных протомеров (субъединиц), которые кодируются геном хромосомы 12, и синтезируется в клеточных структурах эндотелия и мегакариоцитах. Синтезированный в мегакариоцитах ФВ поступает в тромбоциты, хранится в á-гранулах и выбрасывается из последних под влиянием вызывающих агрегацию стимулов. Роль ФВ в гемостазе связана с его мультимерным строением и наличием в каждой из его субъединиц строго дифференцированных доменов связывания с рецептором тромбоцитарной мембраны, коллагеном, фактором VIII и гепариноподобными гликозаминогликанами. В итоге он функционирует как белок – носитель фактора VIII и одновременно опосредует адгезию тромбоцитов к субэндотелиальным структурам поврежденных сосудов. При этом ФВ выполняет роль моста, соединяющего рецепторы тромбоцитарной мембраны Ib и IIb/VIIIa (см. цв. вкл., рис. 5.13 и 5.14), а с другой стороны – коллаген, миофибриллы эластина, гепариноподобный гликозаминогликан. Нарушение путей синтеза и секреции ФВ приводит к возникновению различных форм болезни Виллебранда, различающихся и по тяжести, и по лабораторным проявлениям.
Клиническая картина. Болезнь Виллебранда проявляет себя очень рано. У детей в возрасте от года до 5 лет могут быть подкожные геморрагии и кровотечения из слизистых оболочек полости рта и носа. Причиной повышенной кровоточивости могут быть травмы и различные хирургические вмешательства (тонзиллэктомия, аденоэктомия, экстракции зубов). Наряду с этим бывают и самопроизвольные носовые кровотечения. На фоне вирусных инфекций верхних дыхательных путей последние могут стать профузными и приводить к анемии. При появлении менструаций у девочек они могут продолжаться от недели до месяца и носить характер меноррагий. При тяжелом течении заболевания встречаются спонтанно возникающие желудочно-кишечные кровотечения, апоплексии яичников, кровоизлияния в сетчатку глаза, гемартрозы и даже межмышечные гематомы. Гемартрозы, не столь частые при болезни Виллебранда, обычно поражают коленные, локтевые и голеностопные суставы. При этом клинико-рентгенологическая картина гемартрозов похожа на гемофильные.
В целом клиническое течение болезни отличается большим полиморфизмом клинических симптомов, что в первую очередь связано с различным содержанием и характером структурных изменений ФВ плазмы и тромбоцитов. На основании этих критериев выделяют несколько подвариантов или типов болезни. Первый, наиболее часто встречающийся тип, выделяется на основании уменьшенного содержания в крови структурно неизмененного ФВ и полного набора мультимеров. Тип II может быть охарактеризован нормальным или субнормальным уровнем измененного по структуре плазменного ФВ, в мультимерном наборе которого или отсутствуют высокомолекулярные формы (подтип IIА), или имеет место его повышенная аффинность к Ib-рецептору тромбоцитарной мембраны, что обнаруживает себя ускоренной ристомициновой агрегацией тромбоцитов в плазме больного. Наконец, при самом тяжелом и редко встречаемом типе III ФВ не определяется совсем ни в плазме, ни в тромбоцитах. В течении заболевания отмечаются периоды обострений и ремиссий. При этом в ходе взросления больного тяжесть клинических симптомов ослабевает.
Диагноз. Мысль о болезни Виллебранда возникает у врача при наличии в клинике рано возникающего геморрагического диатеза, обнаруживающего одновременно нарушения и тромбоцитарного, и плазменного звена гемостаза. Диагноз подтверждается следующими лабораторными признаками: 1) увеличение первичной длительности кровотечения; 2) снижение ристомициновой адгезивности и агрегации тромбоцитов, устраняемое добавлением ФВ; 3) удлинение активированного парциального тромбопластинового времени (АПТВ); 4) снижение коагуляционной активности фактора VIII; 5) снижение уровня ФВ в плазме (определяется иммуноферментным методом) или нарушение его мультимерной структуры.
Дифференциальный диагноз болезни Виллебранда прежде всего проводят с гемофилией А. Опыт показывает, что наибольшие диагностические трудности возникают при разграничении легких форм болезней, когда столь характерное для болезни Виллебранда увеличение длительности кровотечения и нарушение адгезии тромбоцитов может быть не представлено. С другой стороны, эти лабораторные признаки могут обнаружиться из-за образования антитромбоцитарных антител на фоне частых переливаний крови у части больных с гемофилиями. В этом случае решающее диагностическое значение будет иметь прямое определение содержания антигена и активности ФВ, которые при гемофилии или не отличаются от нормы, или даже повышены.
Лечение. При легких и умеренных кровотечениях у больных с I типом болезни Виллебранда с нормальным содержанием ФВ в тромбоцитах показано медленное (10 – 20 мин) внутривенное введение аналога естественного антидиуретического гормона (десмопрессина) в дозе 0,3 мкг/кг массы тела в 30 – 50 мл изотонического раствора натрия хлорида. При этом повышение ФВ и антигемофильного глобулина регистрируется уже через 30 мин после начала инфузии и достигает максимума через 1,5 – 2 ч. Он удерживается на таком уровне в течение 12 ч, отчего требует повторного введения. Однако лечить десмопрессином более 3 сут не рекомендуется в связи с опустошением последним гранул хранения ФВ в клетках эндотелия и отсюда – с необходимостью синтеза в них новых порций этого фактора.
Для лечения больных с вариантами заболевания, где ФВ в гранулах хранения отсутствует, используют свежезамороженную плазму или криопреципитат, богатый ФВ. При определении гемостатической дозы препарата руководствуются коррекцией прокоагулянтной активности фактора VIII и первичной длительности кровотечения. Эффективность терапии оценивают клинически – по уменьшению интенсивности и продолжительности кровотечений и рассасыванию кровоизлияний. В случае переливания препаратов крови, содержащих и ФВ, и антигемофильный глобулин, уровень последнего увеличивается пропорционально введенному фактору и поддерживается на хорошем уровне. Необходимость в заместительной терапии антигемофильным глобулином возникает только при профузных и длительных кровотечениях. В этом случае фактор VIII вводится внутривенно до остановки кровотечения каждые 24 ч из расчета 20 ед./кг массы тела. Что касается остановки кровотечений из слизистых оболочек полости рта и носа, для этих целей может быть рекомендовано местное использование и тромбина, и гемостатической губки или марли. Прогноз при различных подвариантах заболевания неодинаков. Он более благоприятен при умеренно выраженной форме болезни Виллебранда. В то же время тяжелое течение болезни, нередко наблюдающееся у больных с III типом болезни Виллебранда, может привести к анкилозу суставов и к тяжелой анемии.
Гемофилии
Гемофилии образуют группу наследственно обусловленных заболеваний крови, связанных с нарушением плазменного звена гемостаза. По современным классификациям могут быть выделены гемофилии А, В, С, D и Е.
Гемофилия А – наследственный геморрагический диатез, связанный с нарушенной продукцией антигемофильного глобулина или фактора VIII. Поскольку ответственный за этот белок ген локализован на Х-хромосоме и является рецессивным, гемофилией А в основном болеют мужчины. Женщины же, носители гена гемофилии, имеющие вторую Х-хромосому (гетерозиготы), этим заболеванием не страдают. Исключение составляют лишь девочки-гомозиготы, которые родились в семье больного гемофилией отца и матери – переносчика этой дефективной Х-хромосомы. Кроме того, у 10 – 15 % больных с приобретенной в ходе жизни мутацией гена антигемофильного глобулина гемофилия А может иметь и ненаследственный характер.
Патогенез. Нарушение гена, ответственного за синтез антигемофильного глобулина, чаще всего ассоциируется с продукцией функционально неполноценного фактора VIII (гемофилия А+) или, реже, с его количественным дефектом (гемофилия А–). В любом случае снижение активности фактора VIII приводит к резкому замедлению свертывания крови по внутреннему пути. При этом степень дефицита фактора VIII тесно связана с выраженностью геморрагического диатеза, что находит отражение и в классификации болезни. В частности, при тяжелой форме гемофилии А активность фактора VIII не превышает 5 %, при средней тяжести течения она варьирует от 5 до 10 %, а при легкой – превышает 10 %.
Клиническая картина. Основным симптомом в клинической картине являются периодически повторяющиеся эпизоды кровоточивости. Как правило, они регистрируются в раннем возрасте, но могут появиться и позднее 20 лет. Несмотря на рождение с геном гемофилии, новорожденные дети кровоточат редко, в том числе после перерезки пуповины. До полугода склонность к кровотечениям выявляется у немногих, и они могут появиться при надрыве уздечки языка или подрезании ногтей. Прорезывание молочных зубов проходит без кровоточивости. Однако острые края самих молочных зубов могут быть причиной кровотечений из-за прикусов языка, щек и губ. Когда ребенок начинает вставать и пробует ходить, возможность травматизации резко возрастает. Из-за того, что он падает, появляются гематомы туловища и головы. Когда же ребенок начинает активно ходить и бегать, открываются возможности для кровоизлияния в суставы, прежде всего коленные, локтевые и голеностопные. «Спонтанные» кровотечения (вне травматизации), прежде всего, присущи тяжелым формам гемофилии. С другой стороны, оперативные вмешательства, даже экстракция зубов, обычно сопровождаются повышенной кровоточивостью. Поскольку образование первичной тромбоцитарной пробки при гемофилии не нарушено, эти постоперационные кровотечения у больных гемофилией возникают не сразу, а через несколько часов, но из-за отсутствия выпадения фибрина могут продолжаться днями и даже неделями.
Как уже отмечено ранее, тяжесть клинических проявлений гемофилии будет зависеть от уровня фактора VIII. Когда он достаточно высок, вероятность «спонтанных» кровотечений минимальна. Наоборот, «спонтанные» кровотечения, в том числе кровоизлияния в суставы, становятся частыми при падении уровня фактора VIII ниже 5 %. Немаловажно и то, что первое кровоизлияние предрасполагает к повторным в тот же сустав.
Поскольку с возрастом тяжесть и распространенность поражений суставов прогрессирует, это может привести к тяжелой их дисфункции и в конце концов к инвалидизации больного. Не менее опасны для больных гемофилией также обширные и напряженные подкожные, межмышечные, субфасциальные и забрюшинные гематомы. Как правило, они болезненны, напряжены, часто флюктуируют, ассоциируются с развитием анемии, подъемом температуры тела и нейтрофильным лейкоцитозом, что симулирует флегмону. Следствием таких гематом может быть сдавление окружающих тканей и сосудов с последующим их некрозом, развитием параличей, нарушением чувствительности и атрофией мышц.
Особо опасными считаются обширные гематомы в мягких тканях подчелюстной области, шеи, зева и глотки, которые могут привести к стенозу верхних дыхательных путей и даже асфиксии.
Диагноз. Мысль о возможности у больного гемофилии возникает у врача при наличии в клинике сцепленного с полом геморрагического диатеза. Этот тип наследования характерен как для гемофилии А, так и для гемофилии В (дефицит фактора IX), которые могут быть окончательно разграничены только лабораторно.
Характерным для гемофилии является нарушение показателей, связанных с оценкой внутреннего пути свертывания крови, т. е. увеличение времени свертывания венозной крови, увеличение времени рекальцификации плазмы и АПТВ наряду с нормальными показателями протромбинового и тромбинового времени. При этом остаются неизмененными тесты на нарушение тромбоцитарного звена гемостаза, как то: число тромбоцитов, длительность кровотечения, а также возможности адгезии и агрегации тромбоцитов. Решающим же в постановке диагноза гемофилии А будет обнаруженное в крови уменьшение коагуляционной активности фактора VIII.
Дифференциальный диагноз тяжелых форм гемофилии А прежде всего следует проводить с гемофилией В, при которой клиническая картина геморрагических проявлений и тип наследования могут быть идентичными. Помогает постановке диагноза использование заменно-перекрестных проб. В частности, коагуляционный дефект при гемофилии А может быть скорригирован плазмой, абсорбированной BaSO4, а при гемофилии B – простой сывороткой. В отличие от болезни Виллебранда, при которой тоже имеет место снижение активности фактора VIII, при гемофилии А не увеличивается время кровотечения и нет нарушения агрегации тромбоцитов с ристомицином.
Лечение. Срочной госпитализации подлежат больные с желудочно-кишечными, почечными, легочными кровотечениями, кровотечениями при прикусе языка, надрыве уздечки верхней губы, гематомами головы, шеи, забрюшинными гематомами, обширными гемартрозами, а также ранениями, требующими наложения швов.
Остановка кровотечения у больных гемофилией А связана с устранением имеющегося дефицита фактора VIII. С этой целью лучше всего использовать концентраты антигемофильного глобулина или криопреципитат плазмы. Для остановки небольших кровотечений допустимо введение свежезамороженной плазмы, которая не позволяет поднять содержание фактора VIII выше 20 %. При этом следует помнить, что из-за опасности гиперволемии предельно допустимый объем вводимой плазмы не должен превышать 25 мл/кг/сут. В то же время свежеконсервированная кровь, содержащая низкое количество фактора VIII (всего 0,3 ед./мл), для лечения гемофилии А малопригодна. Поскольку необходимая активность фактора VIII после трансфузии сохраняется только в течение 12 ч, для полной остановки кровотечения он должен вводиться повторно. Наконец, немаловажно и то, что характер и доза препарата должны быть тесно увязаны с интенсивностью кровотечения или объемом ожидаемого оперативного вмешательства, а планируемый уровень подъема фактора VIII обусловлен локализацией и выраженностью геморрагии.
В частности, для остановки умеренных кровотечений в мышцы без сдавления жизненно важных органов, кровотечений из слизистых полости рта, носа, умеренных гемартрозов, при удалении зуба, исключая моляры, уровень фактора VIII достаточно повысить до 10 – 20 %. Более тяжелые и обширные кровоизлияния в мышцы, тяжелые гемартрозы, желудочно-кишечные кровотечения и кровотечения при удалении нескольких зубов требуют повышения фактора VIII до 20 – 40 %. Наконец, при серьезных хирургических вмешательствах и тяжелых травмах оправданно повышение фактора VIII в первые сутки до 50 %, а в последующие – до 30 %.
Вследствие повторных трансфузий препаратов крови у некоторых больных гемофилией (5 – 10 %) могут сформироваться антитела к фактору VIII, ослабляющие его активность, что ставит врача перед необходимостью повышения общей дозы вводимых препаратов.
Что касается остановки кровотечений из поврежденной кожи и кровотечений из слизистых полости рта и носа, для этого могут быть использованы, с одной стороны, криопреципитат, с другой – местная обработка кровоточащего участка тромбином, тампон с 5 % раствором эпсилонаминокапроновой кислоты, давящая повязка и т. д. При необходимости наложения швов, которые для больного гемофилией могут быть серьезным травмирующим моментом, эта процедура должна проводиться под прикрытием гемостатических средств.
Поскольку основными причинами кровотечений у больного гемофилией могут быть травмы, сам ребенок и его родители должны быть предельно осторожными. В частности, такому больному нежелательно посещение дошкольных детских учреждений и участие в активных играх со сверстниками, езда на велосипеде. С другой стороны, крайне важно занятие плаванием, поскольку в воде опасность кровотечений невелика, а движения в суставах осуществляются легче. Неплохо также с ранних лет прививать такому ребенку интерес к настольным играм, к чтению книг и работе с компьютером, формируя таким образом наклонность к умственному, а не к физическому труду.
Умеренно выраженная гемофилия А обычно протекает без серьезных осложнений. Тяжелые кровотечения наблюдаются лишь в результате операций, повреждений и травм. Напротив, при тяжелой форме заболевания имеют место частые кровотечения, особенно в суставы, что приводит к их необратимым деформациям и инвалидизации больного.
Гемофилия В (болезнь Крисмаса) – заболевание наследственной природы, которое обусловлено нарушением синтеза фактора IX. Заболевание передается по рецессивному типу и, так же как гемофилия А, сцеплено с X-хромосомой.
На его долю приходится от 8 до 15 % всех случаев гемофилий.
Патогенез. Снижение функциональной активности фактора IX может быть связано или с угнетением его синтеза, или с продукцией неполноценных молекул. В любом случае происходит замедление свертывания крови по внутреннему пути, что приводит к нарушению образования вторичной коагуляционной пробки и отсюда к повышенной кровоточивости.