



Коллектив авторов
Внутренние болезни. Том 2
Дифференциальный диагноз следует проводить с другими гемолитическими анемиями наследственного характера, в частности с эритроцитопатиями. В отличие от микросфероцитоза и эллиптоцитоза, в мазках крови больных СКА присутствуют характерные серповидные эритроциты, которые первым двум патологиям не свойственны. Кроме того, для СКА характерны периодически возникающие боли в различных участках тела, нередко заканчивающиеся инфарктами, а также тяжело протекающие инфекции. Вместе с тем для окончательной верификации СКА и ее подвариантов крайне желательно определение гемоглобина Hb S по перечисленным выше методикам.
Лечение. Хотя радикального лечения СКА пока не придумано, основные симптомы заболевания могут быть устранены или смягчены при увеличении объема выпиваемой жидкости, использовании анальгетиков, антибиотиков и, в случае необходимости, заместительной терапии донорскими эритроцитами в объеме, обеспечивающем поддержание гемоглобина крови на уровне 100 г/л. При этом доля Нb S должна составлять < 50 %.
Поскольку увеличение в крови фетального гемоглобина, имеющего более низкое сродство к кислороду, уменьшает выраженность тканевой гипоксии, то назначение препаратов, увеличивающих количество Hb F у больных серповидно-клеточной анемией, можно рассматривать как компонент патогенетической терапии. С этой целью используют: 1) гидроксимочевину (0,15 – 0,3 мг/кг), которая увеличивает содержание Hb F путем торможения рибонуклеотид-редуктазы;
2) 5-азацитидин (гипометилирование ДНК – увеличение экспрессии гена ã-глобина и, отсюда, увеличение Hb F); 3) децитабин (гипометилирование ДНК с увеличением экспрессии гена ã-глобина и нарастанием Hb F); 4) аргинин бутират (ингибитор гистондеацетилазного комплекса); 5) эритропоэтин.
Важным компонентом лечения СКА является профилактика развития гемолитического криза с учетом перечисленных выше факторов риска. В этом плане особое значение имеет профилактика инфекций. В то же время профилактика тромбозов до конца не разработана, поскольку за их генез при СКА ответственны не только гиперкоагуляционный синдром и нарушение реологических свойств крови, но и изменение антитромботической и адгезивной функции эндотелия. По-видимому, более предпочтительно в этой ситуации назначение дезагрегантов, чем прямых антикоагулянтов, хотя в период гемолитического криза может быть оправданно также применение низкомолекулярных гепаринов. Поскольку устранение наблюдающегося во время гемолитического криза болевого синдрома может предотвратить нежелательный для СКА спазм периферических сосудов, уменьшить тканевую гипоксию и дальнейшее нарастание гемолиза, оно патогенетически значимо. С этой целью обычно используют как наркотические, так и ненаркотические анальгетики, вазодилатирующие препараты, антикоагулянты, дезагреганты, гидратацию и ингаляцию кислорода.
При тяжелом течении СКА возможно выполнение аллогенной трансплантации костного мозга, эффективность которой составляет около 70 %.
В качестве профилактики гемосидероза должны применяться хелаторы железа под контролем уровня ферритина. Так как у больных на фоне гемолиза и активного эритропоэза имеет место повышенное потребление фолиевой кислоты, показано профилактическое применение фолиевой кислоты в дозе 2 – 3 мг/сут.
Апластические кризы, которые, как правило, развиваются у детей после парвовирусной инфекции, могут потребовать адекватных трансфузий эритроцитов. В этом случае восстановление гемопоэза обычно происходит спонтанно в течение 10 – 14 сут. Вместе с тем следует помнить, что длительная заместительная терапия донорскими эритроцитами может привести к аллосенсибилизации пациентов, что диктует врачам необходимость применения эритроцитарных сред с учетом индивидуального подбора донора, а также использование фильтрованных, отмытых или размороженных эритроцитов.
5.5.2. Приобретенные гемолитические анемии
Эту довольно обширную группу ГА представляют заболевания как иммунной, так и неиммунной природы. Первые связаны с выработкой в организме агглютинирующих антител различных классов к эритроцитам больного, вторые – с повреждающим действием химических и физических факторов окружающей среды, а также клапанных и сосудистых протезов.
В группу иммунных ГА входят заболевания, при которых разрушение эритроцитов происходит под влиянием антител. Среди них выделяются изоиммунные, трансиммунные, гетероиммунные и аутоиммунные анемии.
В то же время по характеру повреждающих эритроциты антител иммунные гемолитические анемии могут быть: а) с неполными тепловыми агглютининами; б) с тепловыми гемолизинами; в) с полными холодовыми агглютининами; г) с двуфазными гемолизинами, причем появление в крови больного агглютининов способствует развитию внесосудистого гемолиза, а гемолизинов – внутрисосудистого.
Клинический опыт показывает, что наибольший удельный вес в группе приобретенных иммунных ГА (до 97 %) приходится на анемии с неполными тепловыми агглютининами. В большинстве случаев гетероиммунных гемолитических анемий к выработке антител приводят инфекции или лекарства, которые в этом случае выполняют роль провоцирующего синтез антител гаптена. В случае же аутоиммунных ГА ведущее место в патогенезе гемолитического синдрома имеет срыв исходной толерантности лимфоцитов к собственным клеткам организма.
Иммунная гемолитическая анемия с тепловыми агглютининами. Эта форма ГА может начинаться по-разному. Реже имеет место очень бурный процесс, чаще – подострый и даже хронический. В первом случае на фоне полного благополучия у больного неожиданно падает гемоглобин, повышается температура тела и темнеет моча. Причем лихорадка, как правило, тем выше, чем острее протекает сам гемолиз. Селезенка не успевает увеличиться. Костный мозг богат клеточными элементами с расширенным эритроидным ростком. В крови помимо увеличенного количества ретикулоцитов может быть адаптационный лейкоцитоз и выраженный сдвиг лейкоцитарной формулы влево. Нередко встречаются также эритрокариоциты. В других случаях темп развития болезни не столь бурный. Больных больше беспокоят слабость и другие признаки анемического синдрома, хотя может быть зарегистрировано и легкое повышение температуры тела, и увеличение селезенки, и различной степени выраженности желтушность кожи и склер.
Диагноз иммунной ГА с тепловыми агглютининами ставится на основании появления у больного анемического синдрома, не связанного с кровопотерей. Он чаще всего выявляется на фоне какой-нибудь инфекции или приема лекарства, сопровождается разной степени выраженности желтухой, высоким ретикулоцитозом и выраженной адаптационной реакцией эритроидного ростка костного мозга. Гемолиз может быть подтвержден выявлением у больного укорочения жизни эритроцитов и повышением в крови непрямого билирубина. Иммунная природа гемолиза доказывается положительной прямой пробой Кумбса, обнаруживающей наличие фиксированных на эритроцитах больного антител. Если результаты пробы Кумбса будут отрицательны, можно использовать более чувствительный агрегационный тест.
Дифференциальный диагноз иммунной ГА следует проводить с врожденным микросфероцитозом. Поставить окончательный диагноз помогает то, что при микросфероцитозе анемия и связанная с ней желтуха носит отчетливый семейный характер. Она встречается у нескольких членов одной и той же семьи и может сопровождаться отмеченными выше изменениями в костной системе. Кроме того, диагностически значимой в отношении иммунного гемолиза будет положительная проба Кумбса.
Лечение ГА с тепловыми агглютининами начинается с отмены провоцирующих ее лекарств. Неотложная терапия включает назначение глюкокортикоидов из расчета 1 мг/кг массы тела. При отсутствии эффекта начальную дозу преднизолона увеличивают в 1,5 – 2 раза или заменяют его на метилпреднизолон (метипред). При достижении положительного эффекта дозу глюкокортикоидов постепенно снижают. Если на этом фоне наступает рецидив гемолиза, используют ритуксимаб или иммунодепрессанты, в частности азатиоприн (имуран) и сандимун (циклоспорин-А).
Следующей линией обороны считается назначение антилимфоцитарного глобулина и спленэктомия. Положительный эффект операции наблюдается у 65 – 70 % больных, у 15 % урежаются гемолитические кризы, которые теперь проходят без или с уменьшенными дозами стероидов. В то же время у 20 % больных спленэктомия эффекта не приносит. В этих ситуациях могут быть показаны и ритуксимаб, и такие иммунодепрессанты цитостатического действия, как циклофосфан, винкристин (онковин), хлорбутин и альфа-интерферон. В качестве важного стабилизирующего иммунный процесс средства может быть рекомендован также решительный отказ от приема молочных продуктов и животной пищи, которые, как известно, являются мощными естественными аллергенами. К сожалению, несмотря на все эти терапевтические ухищрения, у ряда больных приобретенными иммунными гемолитическими анемиями остановить патологический процесс до конца не удается, что вынуждает больных к длительному приему и глюкокортикоидов, и иммунодепрессантов со всеми вытекающими отсюда последствиями.
Прогноз заболевания достаточно серьезный. Основные осложнения – желчнокаменная болезнь, тесно связанные с терапией глюкокортикоидами различные инфекции, стероидный диабет, язвенная болезнь, артериальная гипертензия, надпочечниковая недостаточность, остеопороз и некоторые другие.
Иммунная гемолитическая анемия с холодовыми агглютининами. Своеобразие этой формы приобретенной гемолитической анемии состоит в том, что она встречается преимущественно у пожилых людей и часто сопровождается резко увеличенной (до 90 мм/ч) СОЭ, которая в термостате «нормализуется». Из других важных клинических и лабораторных признаков должны быть отмечены: выраженная полиагглютинабельность эритроцитов, затрудняющая определение группы крови, наличие в сыворотке крови М-градиента, ложноположительная реакция Вассермана, а также прогрессирующий во времени синдром Рейно.
Диагноз. Мысль о возможности у больного такой необычной формы гемолитической анемии возникает у врача в случае резко увеличенной СОЭ и больших сложностей с определением группы крови. Окончательный диагноз ставится с учетом положительной реакции агглютинабельности донорских эритроцитов I группы в сыворотке крови больного, поставленной в холодильнике, при отсутствии таковой в термостате (37 °C).
Дифференциальный диагноз следует проводить с другими формами гемолитических анемий, в том числе связанных с механическим повреждением эритроцитов. Помогает отличить ее отмеченный выше феномен панагглютинабельности эритроцитов I группы, выраженность агглютинации в холодильнике при отсутствии ее в термостате, а также четкая разница в определении СОЭ при различных температурных режимах.
Лечение. ГА с холодовыми агглютининами на терапию глюкокортикоидами реагирует плохо. Поэтому основной линией обороны считаются иммунодепрессанты, в том числе с цитостатическим действием (имуран, циклофосфан, хлорбутин и мелфалан).
Прогноз заболевания достаточно серьезный. В частности, возможна трансформация в парапротеинемический гемобластоз (множественная миелома).
Иммунная гемолитическая анемия с гемолизинами. При ГА, обусловленной появлением в крови тепловых или, реже, двухфазных гемолизинов, нарушение целостности эритроцитарной мембраны происходит с помощью фиксированного на гемолизине комплемента.
Клиническая картина при ГА с гемолизинами довольно своеобразна. Попадание гемоглобина прямо в кровь, а оттуда в мочу проявляется в клинике повышением температуры тела, болями в пояснице, появлением красной или темной мочи и, редко, присоединением умеренной желтухи. Однако в случае, если свободного гемоглобина в мочу поступает мало или он активно переводится находящимися там клетками в гемосидерин, цвет мочи может быть не изменен.
В анализе крови имеют место анемия разной степени выраженности и ретикулоцитоз. Биохимическое исследование крови в период обострения обнаруживает увеличение свободного гемоглобина и снижение гаптоглобина. Анализ мочи, сделанный в период ее потемнения, обнаруживает присутствие в ней свободного гемоглобина. В то же время при наличии мочевого осадка в анализируемых клетках может быть обнаружен гемосидерин (см. цв. вкл., рис. 5.12). Из других специальных исследований заслуживают внимания проба Кумбса, сахарозный и кислотный тесты, проба на осмотическую стойкость эритроцитов и определение длительности их жизни.
Диагноз ГА с гемолизинами ставится на основании характерной клинической картины и отмеченных выше данных лабораторных исследований.
Дифференциальный диагноз, прежде всего, следует проводить с пароксизмальной ночной гемоглобинурией (болезнь Маркиафавы – Микели) и с другими видами ГА.
В отличие от болезни Маркиафавы – Микели, при которой разрушение патологически измененных и чрезвычайно чувствительных к комплементу эритроцитов происходит в условиях ночного снижения рН крови, при этих видах анемий никакой связи со временем суток нет, положительна проба Кумбса. Помогает постановка сахарозного и кислотного тестов. В основе сахарозного теста лежит способность сахарозы увеличивать связывание комплемента эритроцитами. Для ее постановки берутся 3 пробирки. В первой эритроциты больного смешиваются с сывороткой донора и сахарозой. Во второй пробирке эритроциты донора смешиваются с сывороткой больного и сахарозой. В третьей пробирке эритроциты больного помещаются в собственную сыворотку и сахарозу. При болезни Маркиафавы – Микели максимальный гемолиз будет в первой пробирке, слабее – в третьей и отсутствует – во второй. Объясняется это тем, что при этой патологии резко увеличивается потребление комплемента, и отсюда его содержание будет выше в сыворотке донора, чем самого больного. В то же время неизмененные эритроциты донора в сыворотке больного с пароксизмальной ночной гемоглобинурией не разрушаются. В случае же иммунных анемий гемолиз должен фиксироваться в третьей пробирке, где присутствуют и антитела, и чувствительные к ним эритроциты. В настоящее время выявление клонов пароксизмальной ночной гемоглобинурии достаточно просто осуществляется методом проточной флуорометрии, нацеленной на распознавание дефицитных по анкерному протеину (CD55 и CD59) лимфоцитов.
Что касается разграничения гемолитических анемий с тепловыми и двухфазными гемолизинами, его осуществляют следующим образом. Берут три пробирки с кровью больного. Одну помещают на2чвтермостат, вторую – на такое же время на холод, а третью – на час в термостат и на час – на холод. При наличии в сыворотке крови тепловых гемолизинов гемолиз будет в первой пробирке, а при наличии двухфазных – в третьей. Наконец, гемолиз патологических эритроцитов резко усиливается добавлением к ним слабого раствора соляной кислоты (кислотная проба Хема).
Лечение ГА с тепловыми гемолизинами поначалу не отличается от стандартного. Препаратами выбора являются глюкокортикоиды, доза которых должна быть адекватной, чтобы остановить гемолиз. Положительный эффект терапии оценивается по нормализации температуры. После достижения клинического эффекта, который может быть зарегистрирован у 90 – 95 % больных, доза преднизолона или метипреда постепенно снижается. При этом полная клиническая ремиссия достижима лишь у 5 % больных.
Второй «линией обороны» при гемолитических анемиях с тепловыми гемолизинами является спленэктомия, эффект от которой может быть у 66 – 68 % прооперированных больных. Часть больных после операции снова дают редкие гемолитические кризы, не требующие назначения глюкокортикоидов или купирующиеся ими. У 20 % больных эффекта от спленэктомии достичь не удается, что оправдывает назначение иммунодепрессивной терапии. Согласно накопленному опыту, предпочтительнее использовать циклофосфан или винкристин, чем азатиоприн (имуран), несмотря на то что последний дает значительно меньше осложнений.
5.6. ГЕМОРРАГИЧЕСКИЕ ДИАТЕЗЫ
Геморрагические диатезы представляют собой сложную группу заболеваний, связанных с нарушением сосудистого, тромбоцитарного и/или плазменного звеньев гемостаза. Они встречаются с частотой 10 случаев на 100 тыс. населения. Их лечение требует от врача специальных знаний и большого практического опыта.
механизмом к свертыванию крови является повреждение целостности сосудистой стенки, которое, с одной стороны, активирует адгезию и агрегацию тромбоцитов, с другой (через XII контактный фактор) – запускает внутренний механизм плазменного звена гемостаза (см. цв. вкл., рис. 5.13). Для остановки кровотечения из капилляров и сосудов малого калибра, как правило, достаточно первичной гемостатической пробки из массы адгезированных к коллагену и агрегированных тромбоцитов, в образовании которой принимают также прямое участие фибриноген и фактор Виллебранда (ФВ). Первый обеспечивает контакт тромбоцитов друг с другом, а второй ответствен за контакт тромбоцитов и коллагена.
Основными механизмами остановки кровотечений из более крупных сосудов являются уменьшение их просвета и выпадение фибрина. Фибрин образуется из фибриногена под действием тромбина, который отщепляет от фибриногена препятствующие полимеризации фибринопептиды А и В. В итоге фибриноген превращается в фибрин-мономер. Далее он может трансформировать фибрин-мономерные комплексы, которые под влиянием фибринстабилизирующего фактора XIII превращаются в нерастворимый фибрин. В то же время часть фибрин-мономерных комплексов снова распадается до фибрин-мономеров или поглощается клетками ретикулоэндотелиальной системы.
Классификация геморрагических диатезов:
III. Заболевания сосудов:
1. Наследственные.
2. Приобретенные.
3. Вызванные лекарствами.
III. Нарушения тромбоцитов:
1. Количественные:
а) наследственные;
б) приобретенные.
2. Качественные:
а) наследственные;
б) приобретенные;
в) вызванные лекарствами.
III. Нарушения факторов свертывания крови:
1. Наследственные:
а) один фактор;
б) множественные факторы.
2. Приобретенные: а) один фактор;
б) множественные факторы.
IV. Множественные дефекты системы типа ДВС-синдрома.
Естественными противовесами свертывающей системы крови являются антитромбин III, система протеинов С и S, плазмин и простациклин. Под влиянием гепарина первый способен блокировать практически все основные этапы плазменного звена гемостаза, включая превращение фибриногена в фибрин-мономер. Активированный плазменный протеин С в присутствии протеина S способен расщеплять протеолитически активированные факторы VIII и V и таким образом тормозить образование активного фактора Х и тромбина. Плазмин принимает непосредственное участие в протеолитическом расщеплении нитей фибрина, а при необходимости способен разрушать и другие плазменные факторы свертывания крови. Что касается простациклина, он активно препятствует агрегации тромбоцитов.
5.6.1. Тромбоцитопении
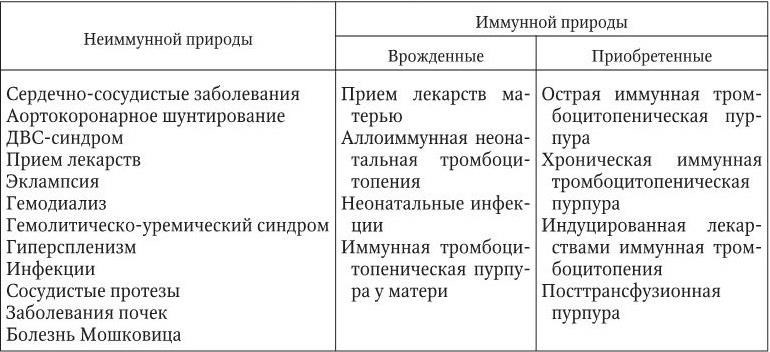
Определение. Под тромбоцитопениями понимаются такие заболевания и синдромы, при которых количество тромбоцитов в крови не достигает 150 % 109/л. Удельный вес тромбоцитопений в группе геморрагических диатезов довольно высок (15 – 20 случаев на 100 тыс. населения). Они могут быть врожденными и приобретенными и вызываются: а) недостаточной продукцией тромбоцитов в костном мозге; б) секвестрацией их в сосудах или в селезенке; в) разведением, в частности, перелитой кровью; г) повышенным разрушением или потреблением тромбоцитов (табл. 5.1).
Таблица 5.1
Основные причины увеличенного потребления или разрушения тромбоцитов
Встречаемость врожденных тромбоцитопений: синдром Алпорта; синдром Бернара – Сулье; врожденный дефицит тромбопоэтина; анемия Фанкони; синдром серых пластинок; аномалия Мея – Хегглина; синдром Вискотта – Олдрича.
Врожденная инфильтрация костного мозга:
а) врожденные лейкемии;
б) врожденный ретикулоэндотелиоз;
в) врожденный мукополисахаридоз;
Врожденная гранулематозная болезнь.
Прием лекарств матерью:
а) этанола;
б) тиазидов;
в) толбутамида;
г) стероидов (эстрогенов или преднизона).
Материнские инфекции:
а) цитомегаловирусная;
б) вируса гепатита;
в) краснухи;
г) оспы.
Иммунные тромбоцитопении (ИТ) представляют сборную группу заболеваний, связанных с выработкой антител к предварительно измененным или даже к неизмененным тромбоцитам. По своей природе они могут быть врожденными и приобретенными, а с учетом механизма образования антител подразделяются на:
а) изоиммунные, или аллоиммунные, при которых разрушение тромбоцитов обусловлено или их несовместимостью по одной из групп крови, или трансфузией реципиенту чужих тромбоцитов при наличии у него ранее выработанных антител к ним;
б) трансиммунные, связанные с переходом к ребенку через плацентарный барьер антител от ранее иммунизированной матери;
в) гетероиммунные, обусловленные первичным нарушением антигенной структуры тромбоцитов вирусом, лекарством или другим агентом, выступающим в роли гаптена;
г) аутоиммунные, при которых из-за срыва толерантности лимфоидной системы лимфоциты начинают вырабатывать антитела к своим исходно неизмененным тромбоцитам.
Как показано в табл. 5.2, важное место среди гетероиммунных тромбоцитопений занимают заболевания, связанные с приемом лекарств. Последние чаще всего выступают в роли гаптена, комплекс которого с иммунными белками плазмы находится на тромбоците, фиксирует комплемент, а затем удаляется из циркуляции макрофагами вместе с несущими иммунные комплексы тромбоцитами. Естественно, что при очередном попадании лекарства в организм сенсибилизированные им больные обнаружат быстрое снижение количества тромбоцитов и, наоборот, их повышение вскоре после отмены препарата.
Таблица 5.2
Лекарства, вызывающие иммунные тромбоцитопении

Аутоиммунная тромбоцитопения представляет собой часто встречающееся заболевание крови. В ее основе лежит образование с последующей фиксацией на тромбоцитах иммуноглобулина G, который и ответственен за их быстрое разрушение в ретикулоэндотелиальной системе.
Недостаток тромбоцитов в крови проявляет себя в нескольких направлениях. Во-первых, не образуется или плохо образуется тромбоцитарная пробка, что приводит к увеличению длительности кровотечения и появлению экхимозов и синяков в местах небольших травм. Во-вторых, из-за недостаточного поступления питательных веществ к эндотелию, главными «кормильцами» которого являются тромбоциты, начинает страдать сосудистая стенка, что, несомненно, способствует и появлению, и прогрессированию геморрагического диатеза.
По течению аутоиммунную тромбоцитопеническую пурпуру можно разделить на острую, рецидивирующую и хроническую формы.
Острая форма болезни более свойственна детям, в равной мере мальчикам и девочкам. Она часто возникает на фоне перенесенных сезонных (зима – весна) вирусных инфекций, в том числе типичных детских инфекций и острых респираторных заболеваний. Промежуток времени между появлением пурпуры и перенесенной инфекцией, как правило, составляет две недели, а средняя продолжительность измеряется 1 – 2 мес. Эта форма благоприятнее, чем хроническая, хотя при отказе от глюкокортикоидной терапии смертельные исходы не считаются редкостью.
Хронической формой преимущественно страдают взрослые, причем женщины в 2 раза чаще мужчин. Нередко заболевание может продолжаться 30 лет и более. Что касается ремиссий, то они в основном заключаются не в нормализации количества тромбоцитов, а в исчезновении геморрагий.
Рецидивирующими формами аутоиммунной тромбоцитопении могут болеть и взрослые, и дети. При этом налицо не только повторные рецидивы заболевания, но и полные ремиссии. В большинстве случаев иммунная тромбоцитопения развивается без отчетливой связи с каким-либо сопутствующим заболеванием. С другой стороны, симптоматические варианты болезни встречаются при хроническом лимфолейкозе, миеломной болезни, хроническом активном гепатите, системной красной волчанке, ревматоидном полиартрите, диффузном токсическом зобе и ряде других заболеваний.
Клиническая картина. Больные обращаются к врачу из-за кровоизлияний в кожу и/или кровотечений из слизистых. Излюбленной локализацией мелкоточечных или петехиальных кровоизлияний является кожа нижних конечностей, где внутрикапиллярное давление выше, чем на верхних. Часть петехиальных кровоизлияний может сливаться, образуя экхимозы. По мере прогрессирования патологического процесса кровоизлияния могут появляться и на туловище, поражать сетчатку глаза и даже мозг. Относительно часто при иммунной тромбоцитопении встречаются носовые кровотечения, гиперполименоррея и кровоточивость десен, особенно после чистки зубов. Характерно также образование синяков в местах инъекций. В то же время кровотечения из желудочно-кишечного тракта, мочевыделительной и дыхательной систем встречаются реже. У части больных иммунными тромбоцитопениями важным клиническим синдромом может быть анемия. В этом случае появляются жалобы на слабость, учащение сердцебиения и головокружение.
Объективное исследование больного выявляет отмеченные выше изменения со стороны кожи и слизистых, может быть анемизация разной степени выраженности. Увеличение селезенки и печени малохарактерно. В то же время у некоторых больных имеет место увеличение лимфатических желез, особенно в области шеи, что в сочетании с субфебрильной температурой тела нередко симулирует системную красную волчанку.
Лабораторная диагностика. Самым простым тестом на тромбоцитопению, который может быть проведен в амбулаторных условиях, является увеличение времени кровотечения. Последнее определяется после нанесения на кожу мелкой раны. Для стандартизации результатов последнюю лучше наносить на средней трети предплечья, в условиях туго наложенной на плечо манжетки от прибора для измерения артериального давления. В норме длительность кровотечения из поврежденного сосуда не должна превышать полутора минут.
Другим важным ориентирующим на тромбоцитопению тестом может быть снижение ретракции кровяного сгустка. Это происходит из-за недостаточного поступления в плазму вырабатываемого тромбоцитами ретрактозима и наглядно проявляет себя невозможностью получения сыворотки из взятой без антикоагулянта свернувшейся крови. В то же время свертываемость крови у таких больных нормальная. Не изменены также каолин-кефалиновый и аутокоагуляционный тесты. Ввиду постоянной циркуляции в крови антитромбоцитарных антител, функциональная активность тромбоцитов больных иммунными тромбоцитопениями может быть изменена, что находит отражение в снижении их способности прилипать к стеклу, а также в нарушении АДФ-, тромбин- и коллаген-агрегации.
Диагноз тромбоцитопении ставится на основании характерной клинической картины и упомянутых выше лабораторных тестов, причем окончательный вывод о наличии у больного такого заболевания делается только после обнаружения в крови уменьшенного (менее 150 % 109/л) количества тромбоцитов. Последние лучше определять вместе с клиническим анализом крови, который сразу же позволит отвергнуть вторичные тромбоцитопении при лейкозах и апластической анемии. Вместе с тем в мазках крови может быть представлено увеличенное содержание мегатромбоцитов. Как следствие повторных геморрагий может развиться гипохромная железодефицитная анемия. Уточнение диагноза требует производства стернальной пункции, которая может обнаружить не только увеличение содержания мегакариоцитов, но и изменение их структуры (гиперсегментация ядра). В качестве вспомогательного диагностического теста может быть использовано определение в плазме и на тромбоцитах больного антитромбоцитарных антител. Если таковые обнаружены, диагноз иммунной тромбоцитопении можно считать доказанным; если нет, то отвергать такой диагноз все-таки преждевременно. Дело в том, что титр указанных антител может быть недостаточным для их идентификации, а наиболее насыщенные антитромбоцитарными антителами пластинки уже захвачены и разрушены макрофагами. Кроме того, следует иметь в виду, что определение антител на поверхности тромбоцитов сопряжено с большими методическими трудностями и поэтому в клинических условиях используется редко.
Дифференциальный диагноз иммунных тромбоцитопений проводят с таковыми неиммунного генеза, а также с симптоматическими тромбоцитопениями. В первом случае помогает поставить диагноз отсутствие у больных иммунными тромбоцитопениями заболеваний сердечно-сосудистой системы, особенно корригируемых постановкой искусственных клапанов сердца или сосудистых протезов, наличие в костном мозге увеличенного количества мегакариоцитов и положительные тесты на антитромбоцитарные антитела. Для исключения лейкозов и аплазий кроветворения учитывают данные клинического анализа крови и соответствующие изменения костного мозга. Для исключения системной красной волчанки, болезни Грейвса и ревматоидного полиартрита учитывают наличие у больных свойственных таким патологиям симптомов и соответствующие лабораторные тесты (LE-клетки, антинуклеарный фактор, антитела к нативной ДНК, ревматоидный фактор, увеличение гормонов щитовидной железы в крови и т. д.).
Лечение. Поскольку некоторые виды ИТ тесно связаны с приемом лекарств или с перенесенной недавно инфекцией, первой задачей лечащего врача является отмена этих препаратов и устранение из организма соответствующего инфекционного агента.
Что касается основных методов специфического лечения ИТ, они следующие:
– глюкокортикоиды;
– иммунодепрессанты (азатиоприн);
– внутривенное введение больших доз IgG;
– антилимфоцитарный глобулин;
– иммунокорректоры (сандиммун);
– спленэктомия;
– малые дозы винкристина или винбластина;
– интерферон-á;
– даназол;
– антитела к СD20 (мабтера);
– ромипластин;
– и другие, реже используемые подходы.
При выборе терапии ИТ учитывают возраст больных. Основным же показанием к ее началу считается наличие у больного выраженного геморрагического синдрома, а не уровень тромбоцитопении.
Начинают лечение с назначения преднизолона в дозе 1 мг/кг веса. Эффект терапии (ослабление или исчезновение геморрагического диатеза, а также увеличение числа тромбоцитов) обычно регистрируется через 5 – 7 дней. При отсутствии эффекта первоначальная доза преднизолона увеличивается в 1,5 – 2 и даже в 3 раза. Показанием для снижения дозы преднизолона (не быстрее, чем 5 мг в 4 дня) будет полная ликвидация геморрагического диатеза на фоне отчетливо наметившейся тенденции к повышению количества тромбоцитов в крови. Если в ходе отмены глюкокортикоидов геморрагический диатез возобновляется или будет зарегистрировано критическое снижение тромбоцитов (ниже 10 % 109/л), терапию преднизолоном заменяют на короткий курс метилпреднизолона (30 мг/кг/сут в течение 5 дней) или подключают к основной терапии азатиоприн (0,05 % 3 раза в день).